
Making batteries more like bombs
What would it take to 100x the power density of electrochemical batteries?

We build batteries as mechanical devices, combining anodes, cathodes, separators, and current collectors microns or mm apart. Could we make them far more powerful by instead synthesizing them as chemical assemblies— by integrating anode, cathode, and separator as separate functional groups along a single polymer chain? That could be the key to constructing ultra-compact thin-film power supplies that have comparable energy density to Li-ion cells, but have vastly higher power density, higher working voltages, and near-infinite cycle lives.
Explosives
Making chemistry go fast is often a matter of getting two reactive materials as close together as physically possible without letting them blow up before you can get out of the way.
Consider explosives—some of the fastest and most energetic chemistry there is. Prior to 1845, the best way to make an explosive was to physically grind up an oxidizer like saltpeter (KNO3) and a reducing agent like charcoal (>C=) to as fine a powder as possible. Blend them together with sulfur that could melt and help them mix, light them, and – poof. Gunpowder suitable for a Revolutionary War musket. A deflagration—but not a detonation.
A key breakthrough to making truly powerful explosives was putting oxidizing and reducing chemicals onto the same molecule. That way instead of having the reducing agent (C) and the oxidizing agent (NO2) microns or even millimeters apart in different grains of material, on compounds like nitroglycerin or TNT the oxidizing and reducing groups were only angstroms apart—thousands or even millions of times closer together. Explosive velocities went from ~100m/s to nearly 10,000m/s. Railroads got built much faster. Wars got more horrible. The world was never the same…
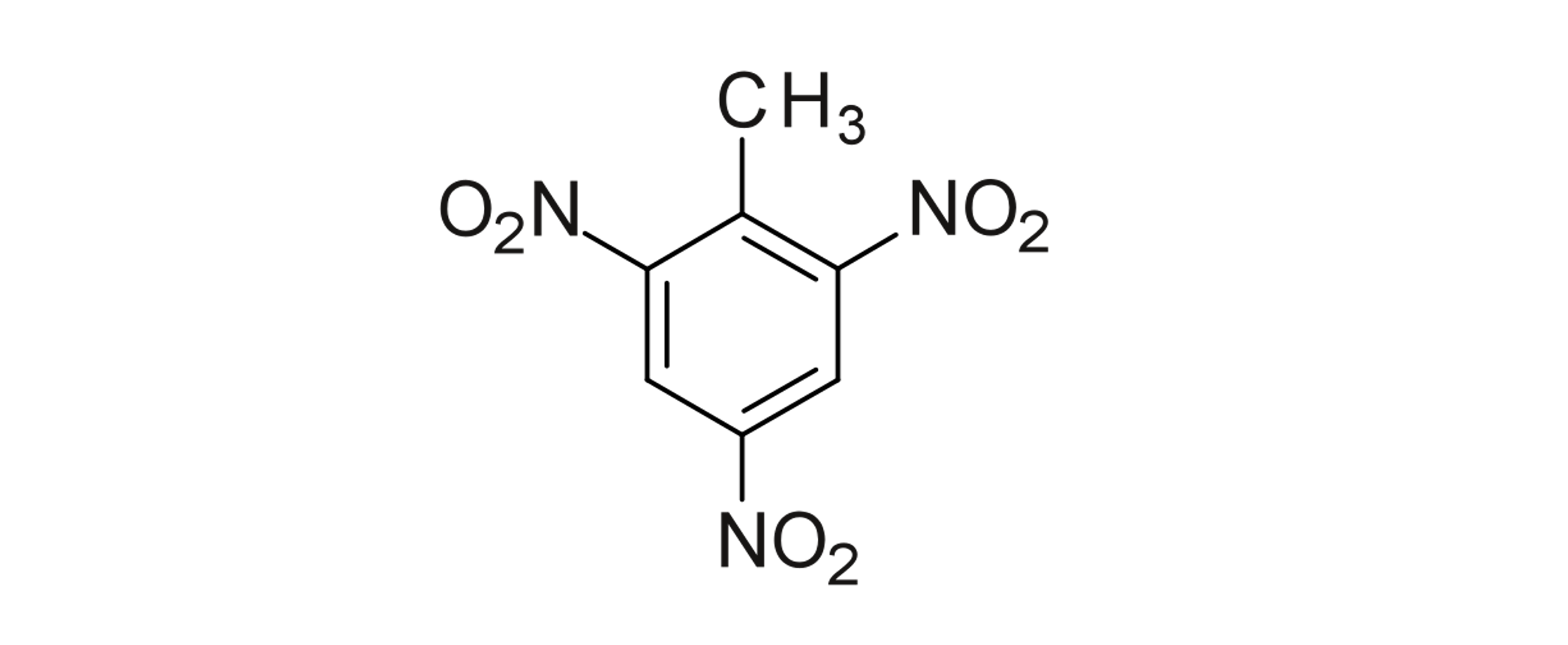
TNT. Megajoules per mol ready to explode with only 1.5 angstroms rearrangement, kept stable by the resonance in benzene.
Batteries
Batteries are a lot like explosives. Like explosives, batteries contain both reducing chemicals and oxidizing chemicals bundled tightly together, ready to react with one another. Like explosives, we engineer our batteries to pack these chemicals together as tightly as possible to speed reaction rates. Like explosives, we like our batteries as powerful as possible…
But the way we put batteries together is a bit like how we made gunpowder in the 1700s. Even though all of the salient reactions and transport phenomena occur on the angstrom-scale, we build batteries in mechanically separated layers—100μm anode, 30μm separator, 100μm cathode etc. Tiny as that seems, that’s as enormous in chemical terms as grains of charcoal and saltpeter in a musket. Even 20 microns leaves tens of thousands of separator molecules for each ion to crawl through before it can cough up an electron into your favorite circuit. So slow!
So here’s the obvious question: what if we could make batteries more like TNT, and assemble them at the molecular scale, with anode and cathode only angstroms apart?
Polymer chemistry seems to give us the tools for this kind of thing fairly straightforwardly. We already have plenty of ion-conducting polymers—that’ll do for the separator. We also have oxidizing and reducing monomers, and conjugated electron-conducting elements. All the ingredients for a tiny battery.
You could start by connecting reversible redox-active end groups with different standard reduction potentials to each end of an ion-conducting polymer. Then the polymer chain becomes a single-electron charge-pump. If you could arrange many such charge pumps on a current-collecting substrate, you’d be looking at an extremely compact power source. This thin-film all-organic system could have mega advantages over traditional multicomponent batteries, which could include huge power density, improved cycle stability (since reactants and separator are covalently attached), and potentially the ability to include huge numbers of cells in series in a single layer of material.
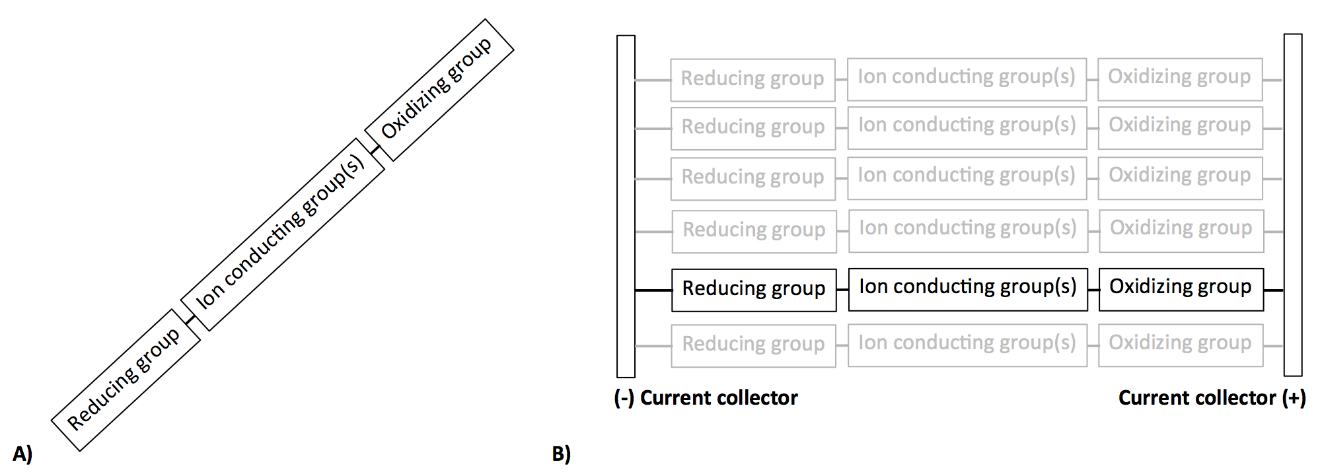
A) single molecule battery and B) an ordered film of single-molecule batteries arranged in parallel between current collectors.
A proof of concept
[TLDR for this section: this seems easy enough in principle—but getting to practical systems might take fancier chemistry than I outline here. ]
Many potential redox active substituents and ion-conductive central chains could be adapted to fit this strategy. Here’s one option I dreamed up by drawing the pathway then googling a bunch of papers where people did each step individually:
Imagine an ion-conducting polyethylene oxide chain (PEO) with anode/cathode made from ferrocene (FC) and anthraquinone (AQ) terminal groups. This chain could be covalently attached to a graphene substrate (or alternately, adsorbed onto Cu or Ni) at the AQ end to form an oriented film. That’s the battery and one contact.
Likely synthesis steps for an FC-AC single molecule battery. Here’s the synthesis. Make monodisperse PEO via living anionic polymerization[1] of ethylene oxide in THF using sodium methyl sulfoxide initiator as described in [ref 4] and elsewhere. Then purify and redissolve it in THF. Separately, get a “positive electrode” ready by preparing ferrocenyl methanol via hydroboration and oxidation of vinyl ferrocene in THF/BH3 and H2O2/KOH. Then replace the sulfoxide end group on the PEO with the FC by combining the PEO-sulfoxide with ferrocenyl methanol and NaH in THF as described in [ref 5] to yield product A, below. This is your separator attached to your positive electrode:
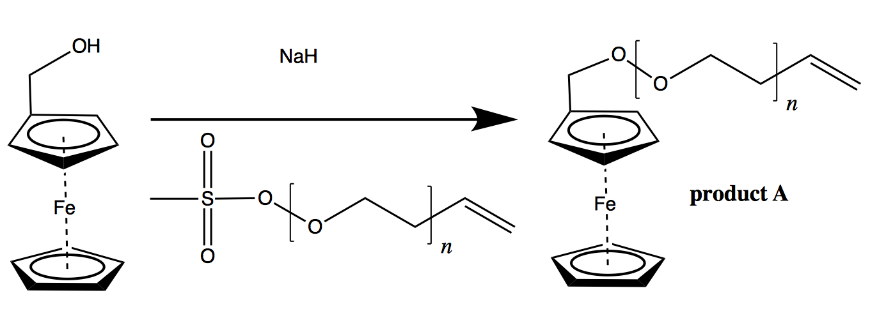
Creating ferrocene-terminated PEO chains by functionalization of PEO oligomers subsequent to living anionic polymerization. This is your positive electrode and separator.
Now let’s work on a negative electrode and current collector. We’re going to attach AQ to a graphene surface using an aryldiazonium coupling reaction. Make a diazonium derivative of AQ (PAQ-N2+) from 2-amino-9,10-phenanthrenequinone (PAQ-NH2) through reaction with (NO)PF6 in acetonitrile at −30 °C, as described in [ref 6]. Subsequently, immerse a graphene substrate in an acetonitrile solution of (PAQ-N2)PF6 and (Bu4N)PF6 to attach the AQ covalently onto the graphitic sp2 sites as in [ref 7].

Covalent attachment of anthraquinone (AQ) onto graphene substrate using an aryldiazonium coupling reaction. This is your current collector and negative electrode.
Now we need to attach the two products A and B. Brominate the terminal vinyl group of product A in a CCl4 mixture, and combine it with product B in methanol with 0.1 equivalents PdCl2 to get a Heck coupling of product A with product B. While this Heck coupling could in theory attack either end of the surface-bound AQ, in all likelihood steric hindrance from the full monolayer of AQ present on the graphene would promote self-assembly of the PEO-FC units into an ordered film.
Finally, you could remove the coupled product (graphene + AQ/PEO/FC film) from the Heck reaction mixture, and put it in a suitable electrolyte. Put on a top electrical contact in order to test the battery, either by metallization or with a conformal conductive carbon paste, to yield the final product, as shown below. Time to touch it with your multimeter and see if it worked.
In a mildly acidic aqueous or alcohol electrolyte, this battery should give you a per-cell potential difference of approximately 300 mV (the potential between 2x-substituted anthraquinone and ferrocene reported in [refs 8, 9]). With use of a Li salt (i.e. LiPF6 in DMC, as shown in Figure 4), this battery would produce approximately 500 mV, based on the reduction potentials shown in [refs 7, 10][2].

The final polymer product between a graphene substrate and positive electrode contact. Touch it with your multimeter and see if it worked.
Getting to high voltages
Obviously less than half a volt isn’t much to write home about. This idea only gets cool when you start to dream up polymeric batteries with multiple cells in series.
Imagine a self-assembled monolayer of polymer chains consisting of four repeating units: a conjugated e- conductor, a reducing agent, an ion conductor, and an oxidizing agent. Arrays of oligomers with tens or hundreds of such units could conceivably be used as a thin-film power source with much higher working voltages than you could get in an ordinary battery.
One technical challenge that would need to be addressed before producing multi-cell systems is getting truly ionomeric conduction: for this concept to produce a high voltage, ion conduction must occur exclusively along the polymer chain, without participation from unbound solvent molecules or significant inter-chain movement (as in our Li-PEO proof of concept battery above) [ref 11]. If ions are allowed to move freely along the length of the polymer, an ionic shunt pathway exists between all the separate redox units along the chain, and you wouldn’t measure a high voltage[3]. There’s probably a way around this (something something pendant groups?) but it's not obvious to me.

A multi-cell, high-voltage oligomeric power supply. Such a system would be assembled into an ordered film of many such units in parallel.
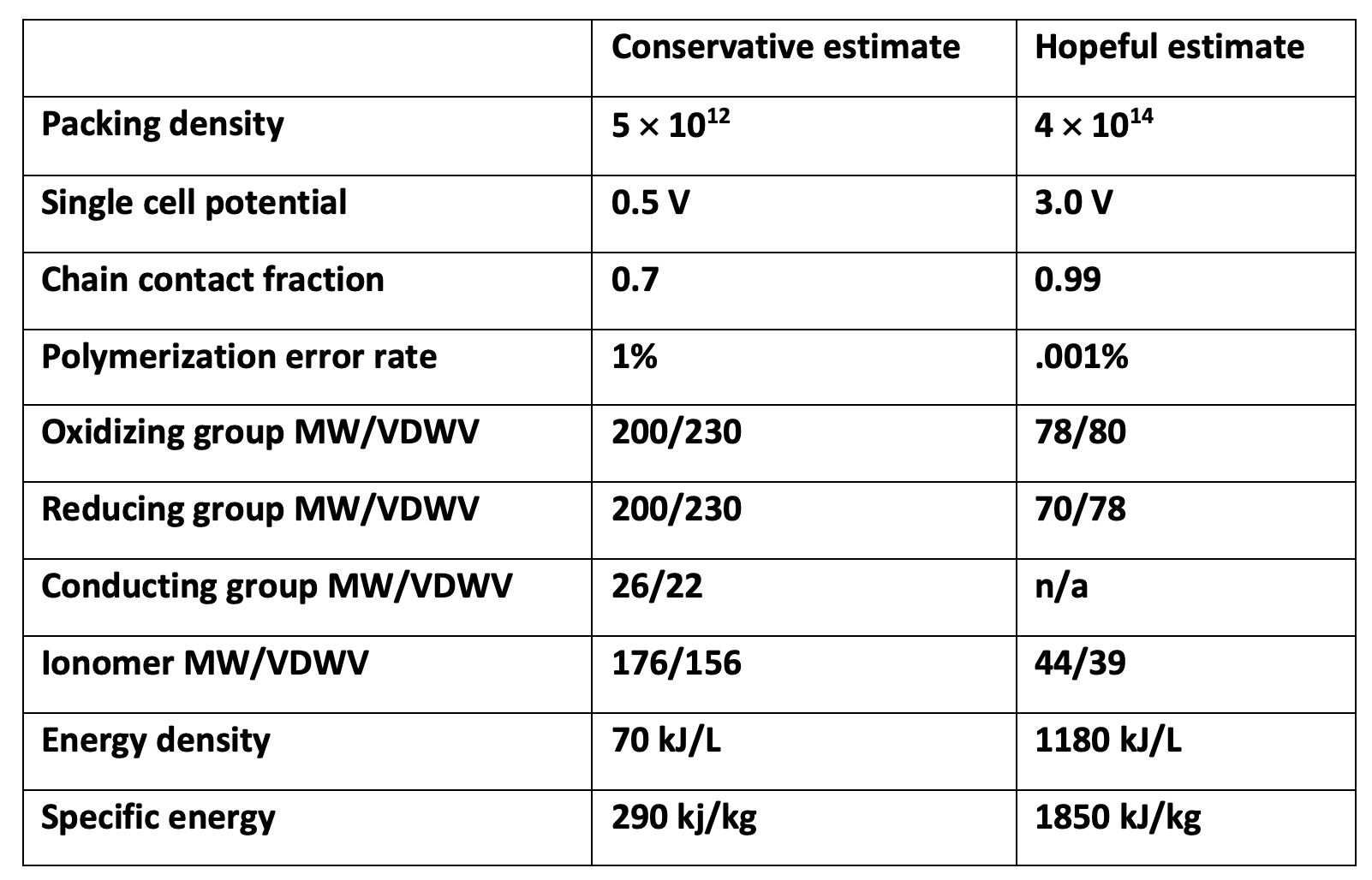
Estimating energy density. The energy density we can get will depend on the polymer chain packing density, the redox potentials of the oxidizing and reducing groups, the fraction of polymer chains making good contact to both current collectors, and whether ‘inert’ polymer strands must be intermixed with the redox-active molecules for structural stability. It’s something of a ‘Drake equation’ until somebody tries this in lab; reasonable estimates range from 100 to 1500 kJ/L. An ordered film prepared with ferrocene and anthraquinone as described above (but linked cell-cell with polyacetylene) might net 250 kJ/L.

Since energy density is so strongly dependent on the polymerization error rate, we need super high polymerization fidelity, especially if we want to get to high voltages. But note that no matter what, this strategy will have lower energy density than Li-ion. In the best case it’d have energy density similar to LFP, maybe half or 2/3rds as good as NMC or LCO. That’s because this battery always has equimolar anode/cathode and separator/conductor, whereas real batteries can be manufactured with much thicker electrodes than separators. The goal here is size and power and reversibility, not energy density.
Estimating the energy density of a TNT-inspired battery. The impact of polymerization error rate on attainable energy density depends strongly on the number of ‘cells’ on each chain, and is given here for a 30 cell (15-90V) film. You could put e.g. foil layers between successive lower molecular weight films to reduce the impact of polymerization errors, but that would reduce energy density and increase manufacturing costs. Molecular weights (MW, amu), and Van der Waal volumes (VDWV, mL/mol) are taken from literature. The specific energy estimate in the conservative case might still be an overestimate, since at such low chain density the polymer would likely require a supporting scaffold between current collectors.
The idea here plays to the strengths of polymer materials, taking advantage of their good dielectric properties and mitigating their relatively lower electronic conductivity by getting to higher voltages. But of course there are a ton of things that could go wrong. You could find you need a whole lot of separator per redox active group. You could fail to crystalize the polymer and have individual “cells” pointing sideways or backwards. If a metal salt is involved, it could migrate along the chains and plate out at one contact. And being polymer materials, the internal resistance could end up being super high, negating the whole high power gambit from the start.
Overall, this seems like a high-risk / high reward idea, and exactly the type of around-the-corner thing that academic battery or polymer chemistry labs should be working on, if they could only get their incentives and signaling sorted. Maybe 10% chance it works at all, 1% chance it changes the world…100% chance you learn something along the way.
For our group though, this chemistry is just too involved. Figuring out whether it has any legs could take years. I could see this taking a PhD’s worth of work to even measure anything at the single-cell scale, before you could even start to talk about engineering anything useful.
The trick with Orca’s other chemistry projects is that they’re dead simple—Savor is like the chemistry of fuels plus the chemistry of making soap, Peregrine is like a PEM electrolyzer with a new reactant piped in at one side. All the pieces have been built before, it’s easy to diligence and show that the TEA works at scale. Both those projects started working almost right away when we built them. This… probably wouldn’t work the first time you tried.
Making batteries like explosives was something I’d been wondering about for years but had never put hard cycles into. Happy to have finished the brainstorm, but clearly it’s far too upstream with the chemistry for us.
References
[1] D. Abagli, Well-Defined Redox-Active Polymers and Block Copolymers , J. Am. Chem. Soc. 114, 4150 (1992)
[2] Kuramoto et al., Ferrocene as an effective initiator for copolymerization of styrene with maleic anhydride, J. Pol. Sci. A 33, 967 (1995)
[3] Denisov et al. Mechanism of Inhibiting Action of Quinones in Oxidizing Polymers and Model Compounds, Macromol. Symp. 143, 65-74 (1999)
[4] Hale et al. Electrical Communication Between Glucose Oxidase and Novel Ferrocene-Containing Siloxane-Ethylene Oxide Copolymers: Biosensor Applications, An. Letters 26, 1-16, (2006)
[5] Ibid, 3
[6] Mahouche-Chergui et al., Aryl diazonium salts: a new class of coupling agents for bonding polymers, biomacromolecules and nanoparticles to surfaces. Chem. Soc. Rev., 40, 4143−4166 (2011)
[7] Jaffe et al., Quinone-Functionalized Carbon Black Cathodes for Lithium Batteries with High Power Densities, Chem. Mater, 27, 3568−3571 (2015)
[8] Huskinson et al., A metal-free organic-inorganic aqueous flow battery, Nature 505, 195 (2014)
[9] Gagne et al., Ferrocene as an internal standard for electrochemical measurements, Inorg. Chem., 9, 19 (1980)
[10] Su et al., Synthesis of a novel ferrocene-contained polypyrrole derivative and its performance as a cathode material for Li-ion batteries, Electrochimica Acta, 104, 302-307, (2013)
[11] Nederstedt et al., Single-ion conducting polymer electrolytes with alternating ionic mesogen-like moieties interconnected by poly(ethylene oxide) segments, Polymer, 177, 231-240 (2019)
[12] Chemical Retrieval on the Web (CROW) Polymer database
Footnotes
[1] While we could make this fairly easily by using a vinyl-ferrocene initiator [ref 1] and AQ terminator [refs 2, 3] in uncontrolled radical polymerization, the high molecular weight (MW) polydispersity that would result from this strategy would bite you, since you need very uniform MW so that as many of the polymer chains as possible are in contact with both electrodes. Otherwise the top electrical contact is a picnic blanket only touching the taller blades of grass. So if you found that you were having trouble making electrical contact to both ends of the supramolecular battery here, you might try living anionic polymerization of PEO, followed by post-functionalization with FC and AQ in order to achieve more uniform MW in the final film.
[2] An attentive reader might notice that the battery based on the Li salt would function at a higher voltage without the quinone unit (i.e., if the FC-PEO chain were attached directly to the graphene, allowing for the more reducing Li-graphite interaction rather than the Li-AQ interaction). However, the graphene redox wouldn’t allow let you build up to multi-cell systems, which is where this concept starts to get cool.
[3] Electrochemical crosstalk between adjacent chains wouldn’t be much of a concern, since substituents at the same coordinate along the polymer chain will be at the same chemical potential. The challenge is to keep ions from moving along the chains.
A proof-of-feasibility might be the operation of chlorophyll molecular networks in the chloroplasts of plants, in which significantly larger electric fields are routinely photo-generated by sunlight and then manipulated in the process of fixing CO2.